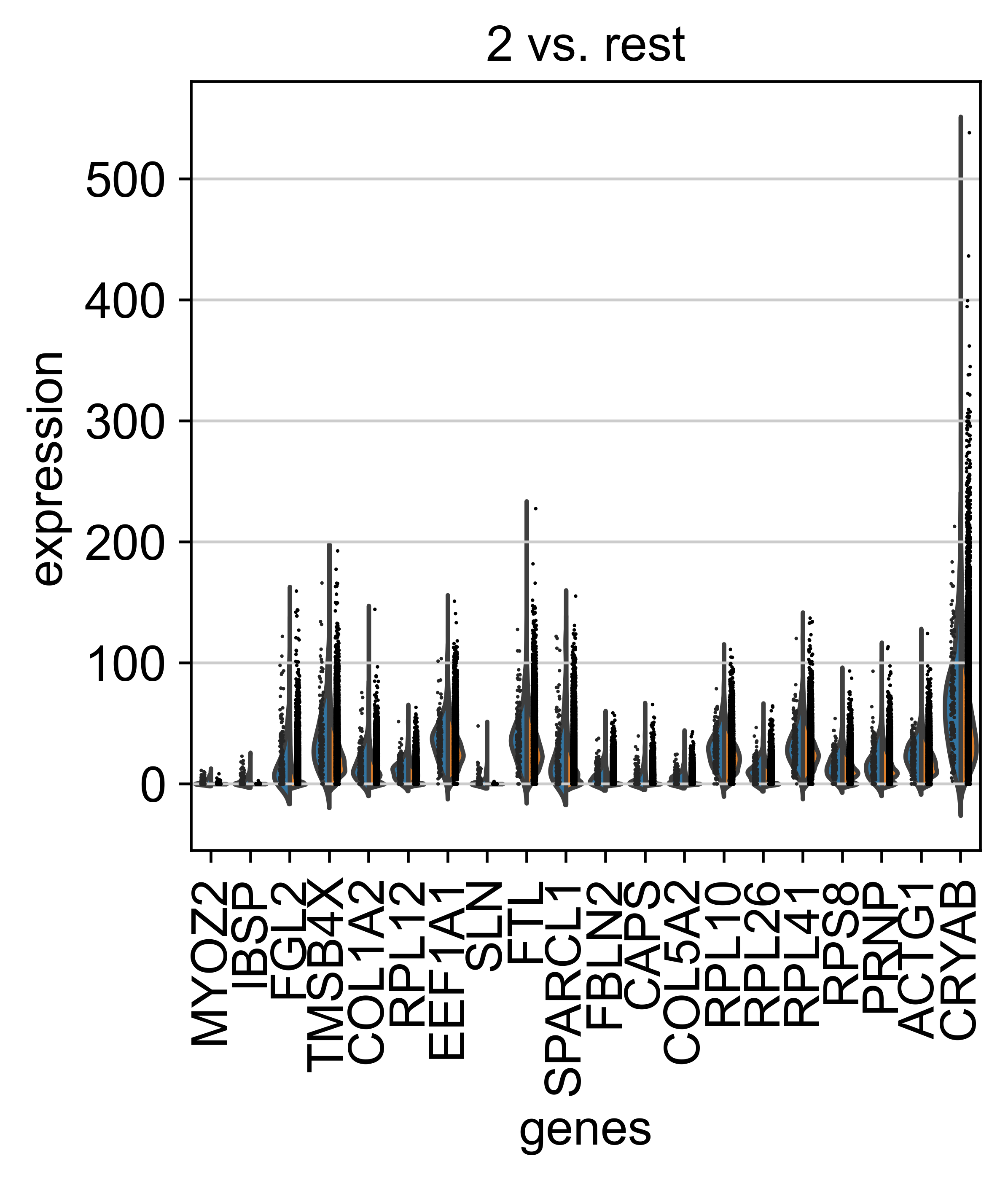
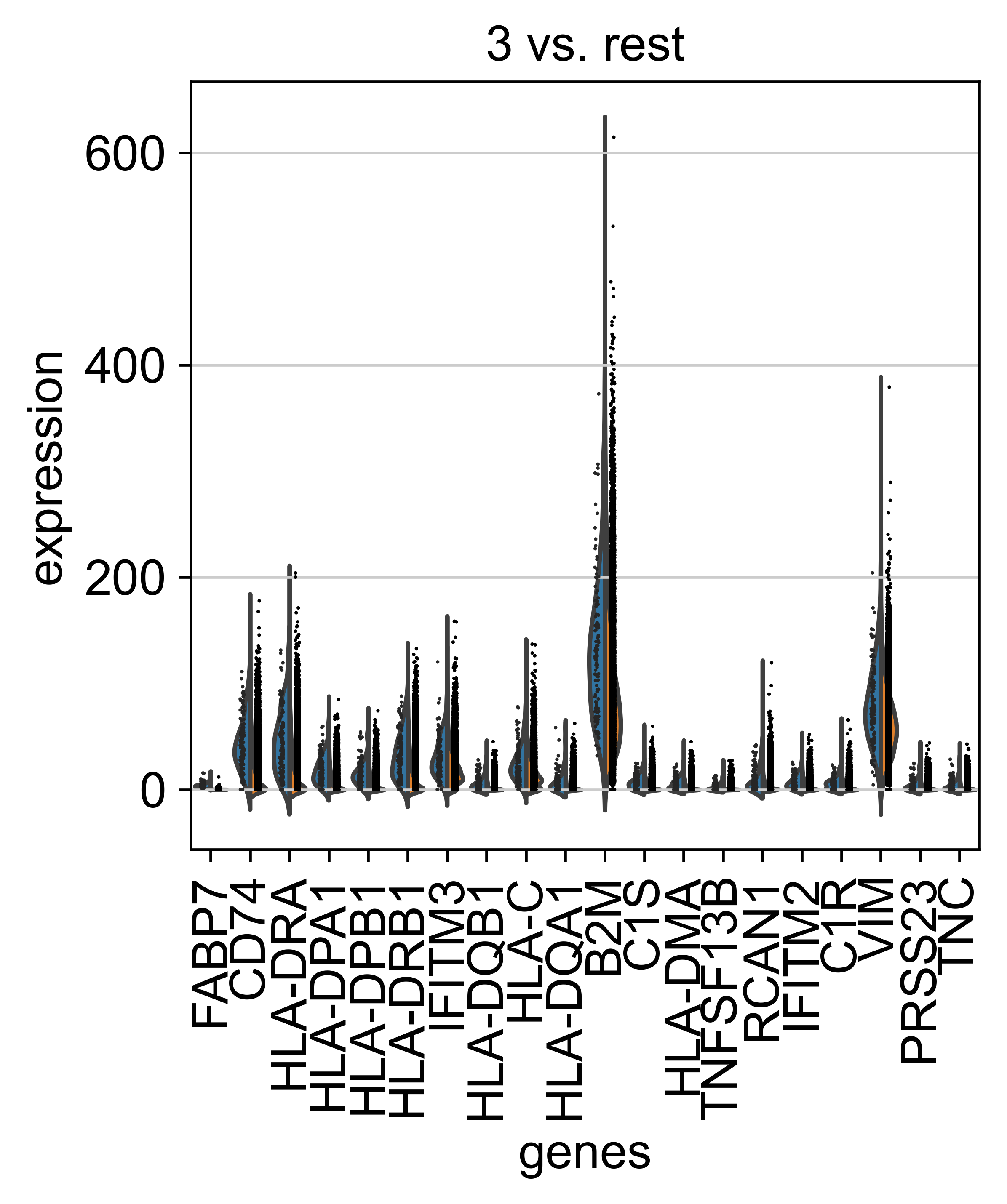
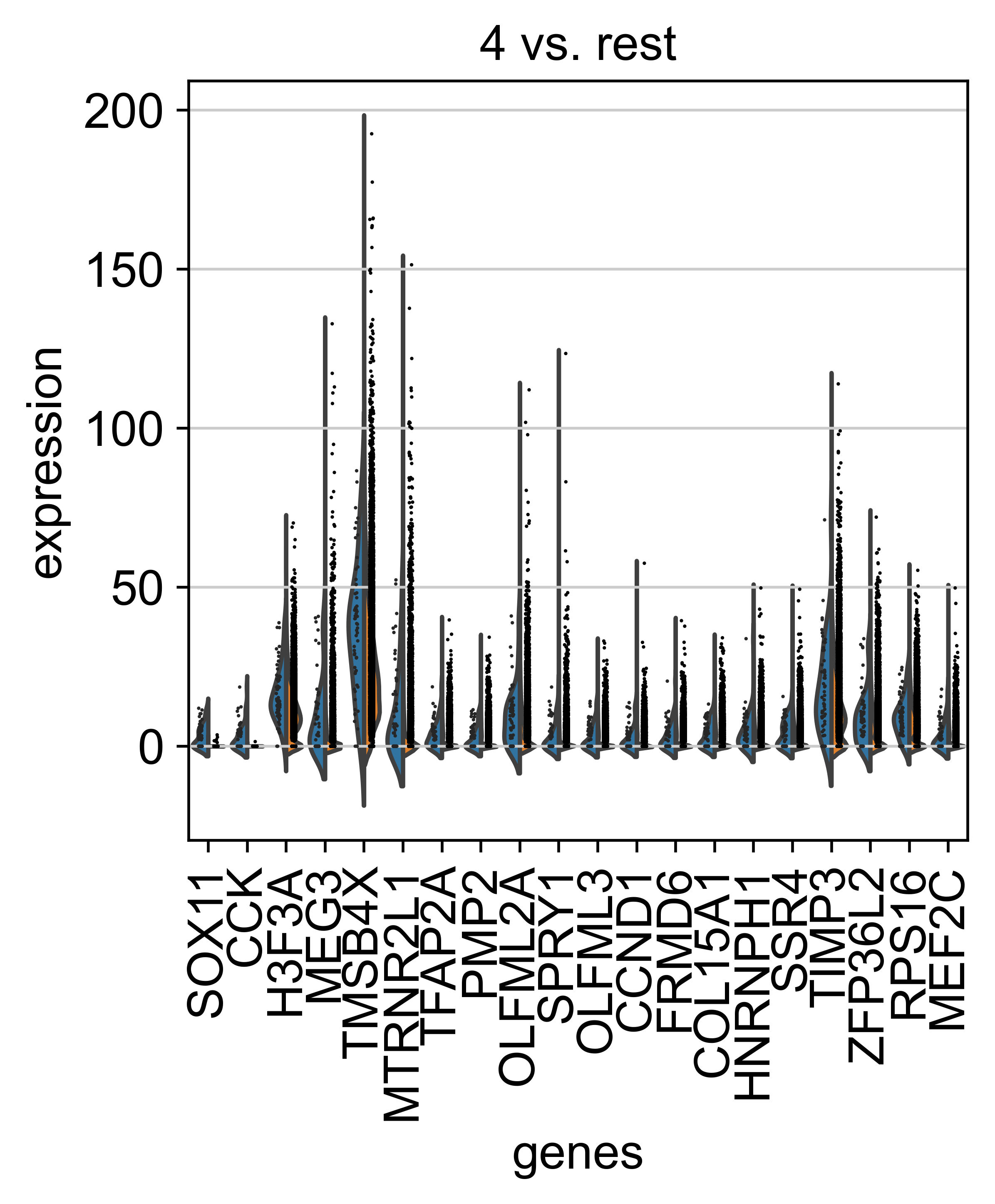
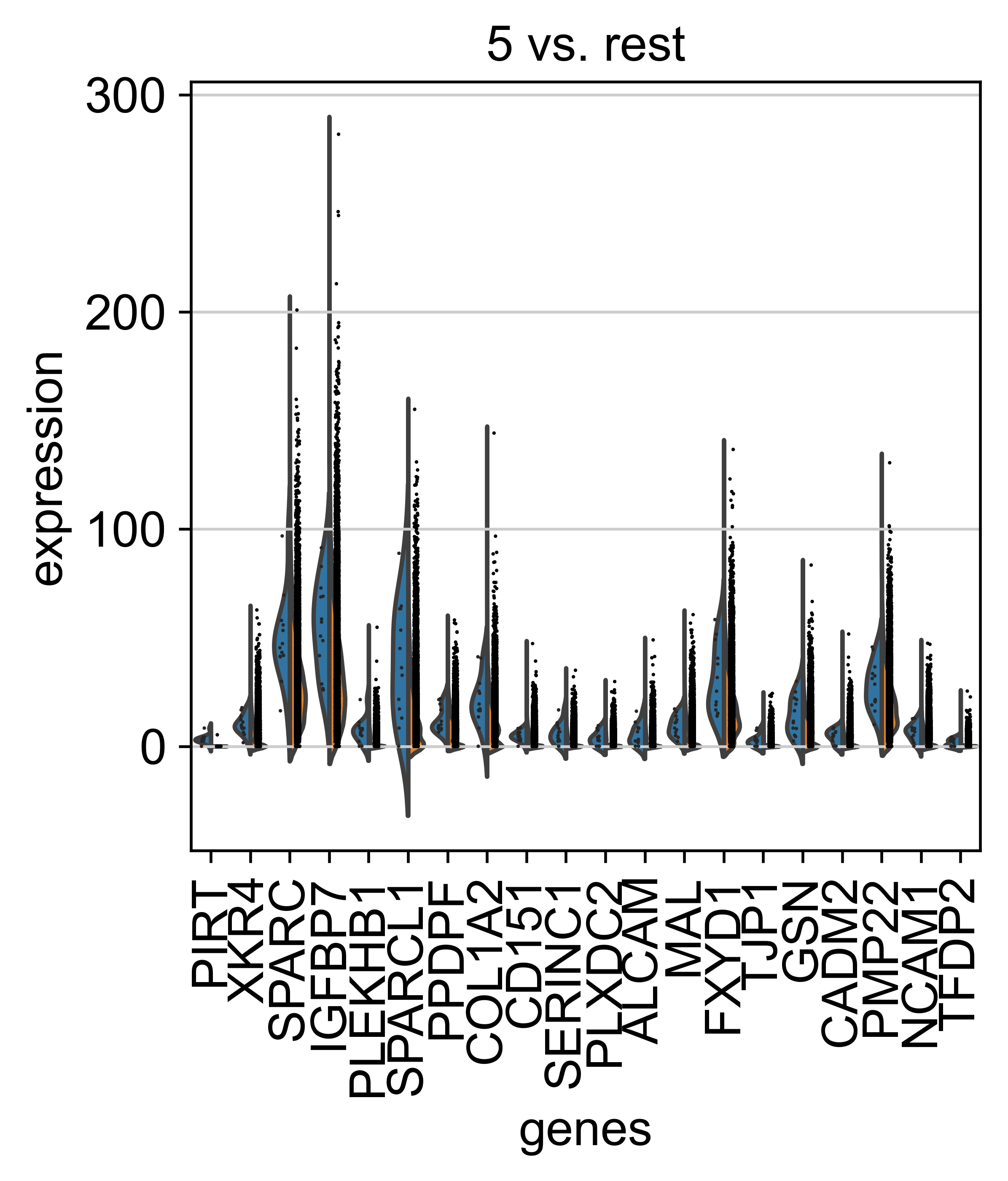
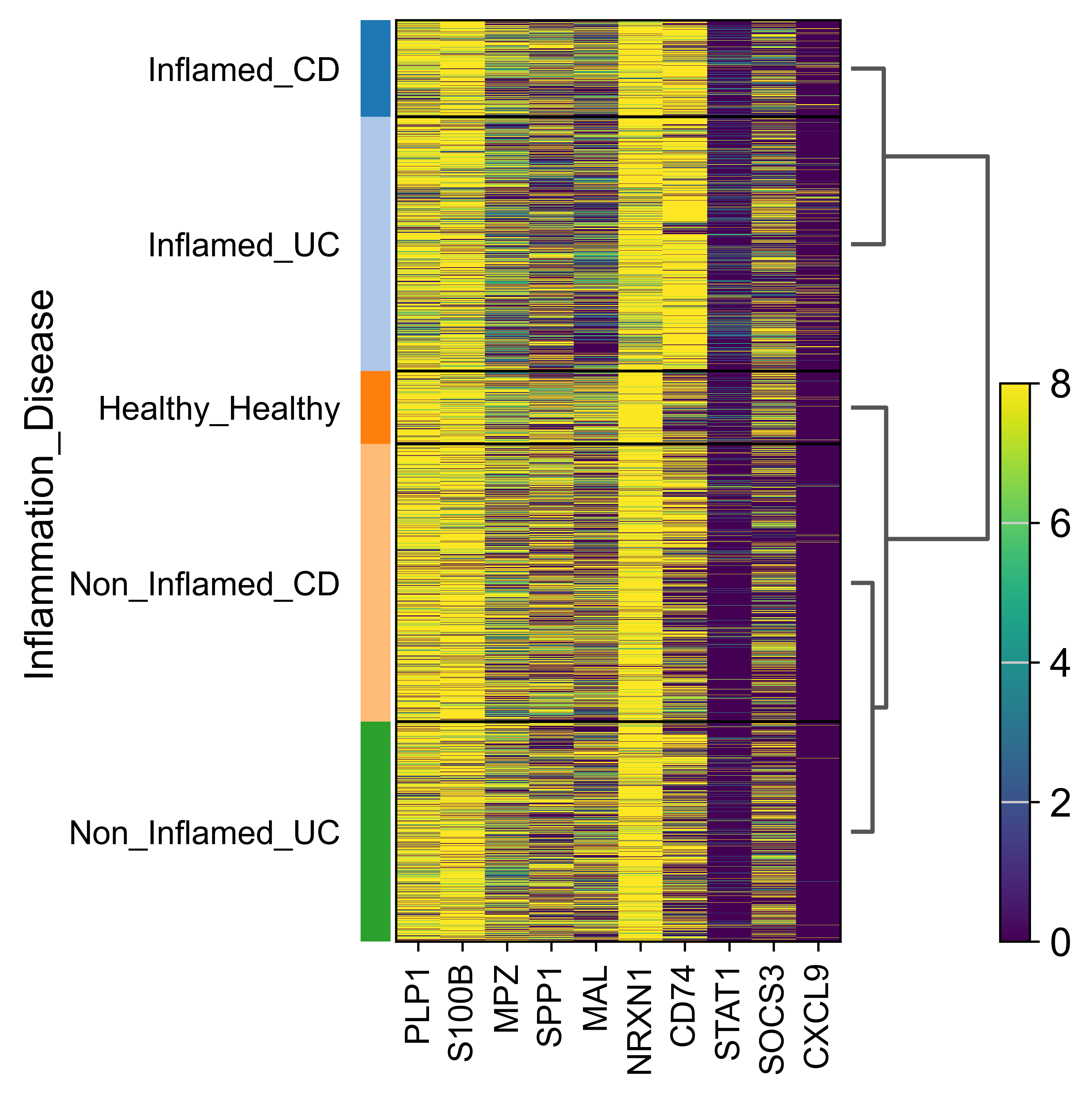
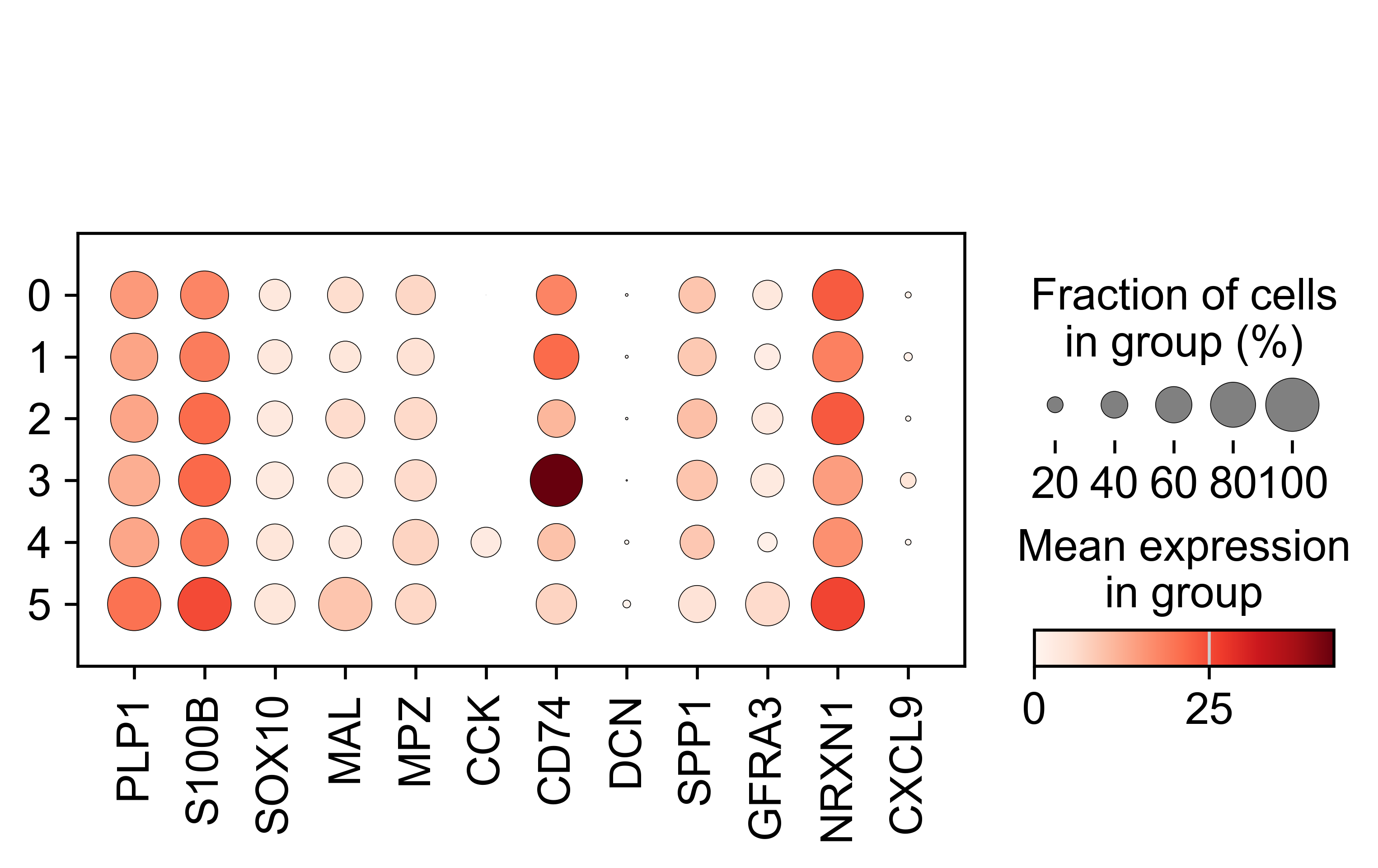
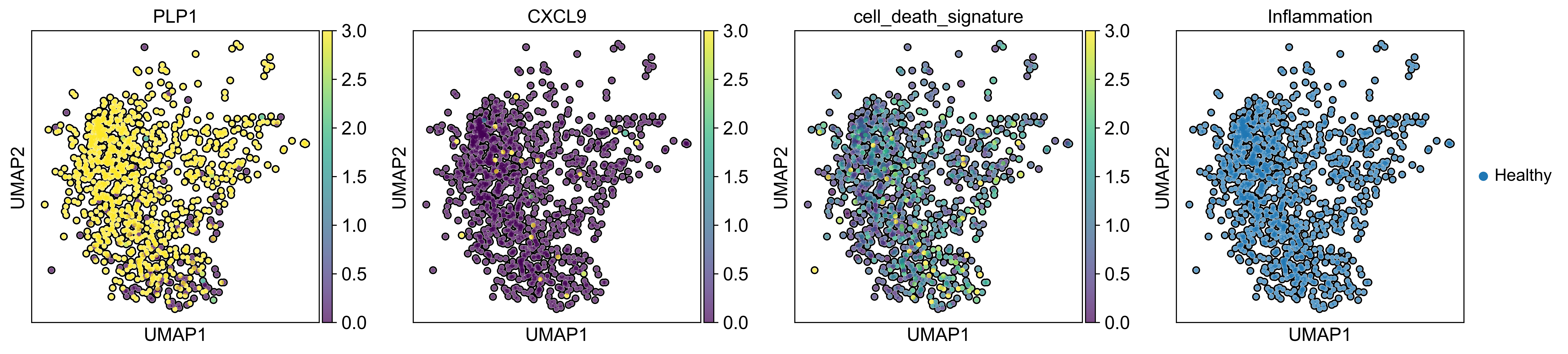
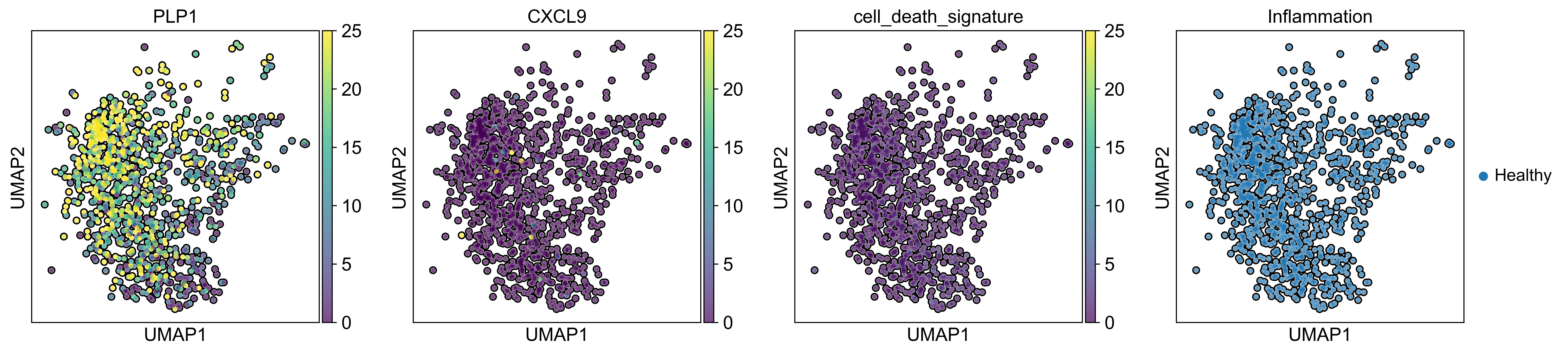
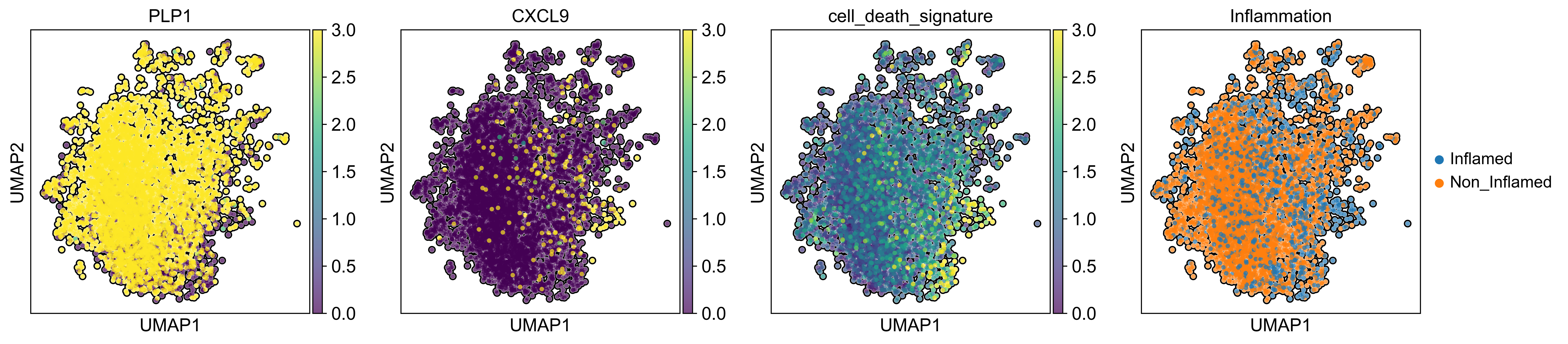
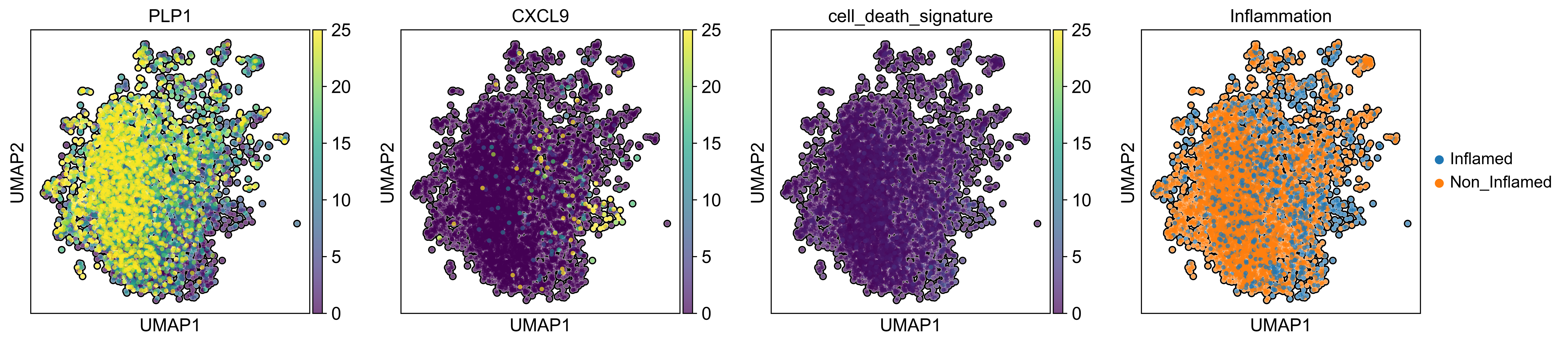
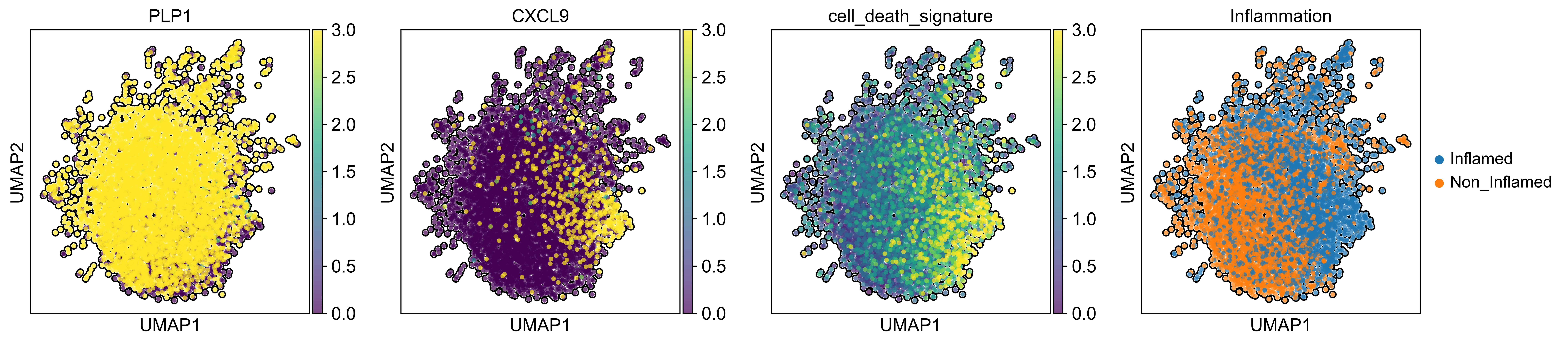
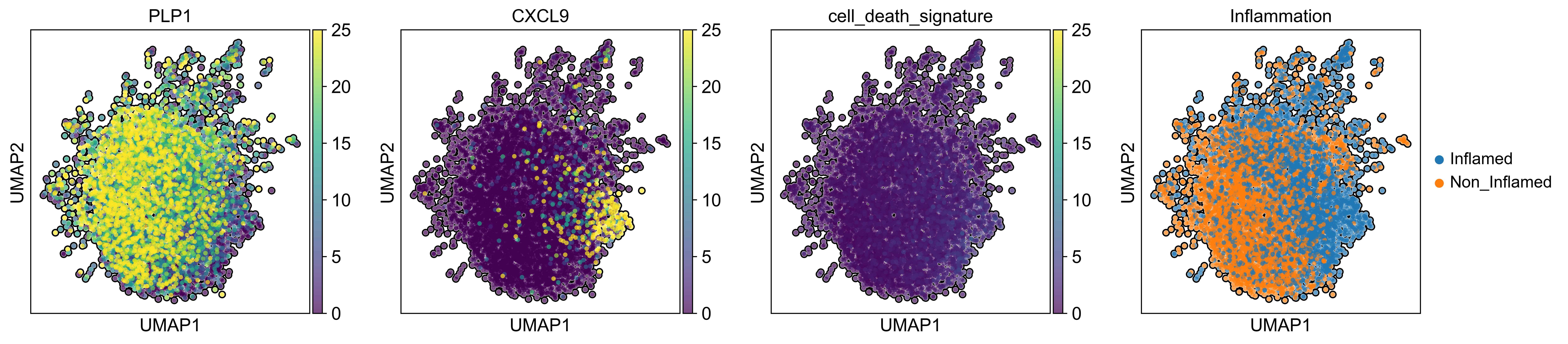
AnnData object with n_obs × n_vars = 71809 × 33075
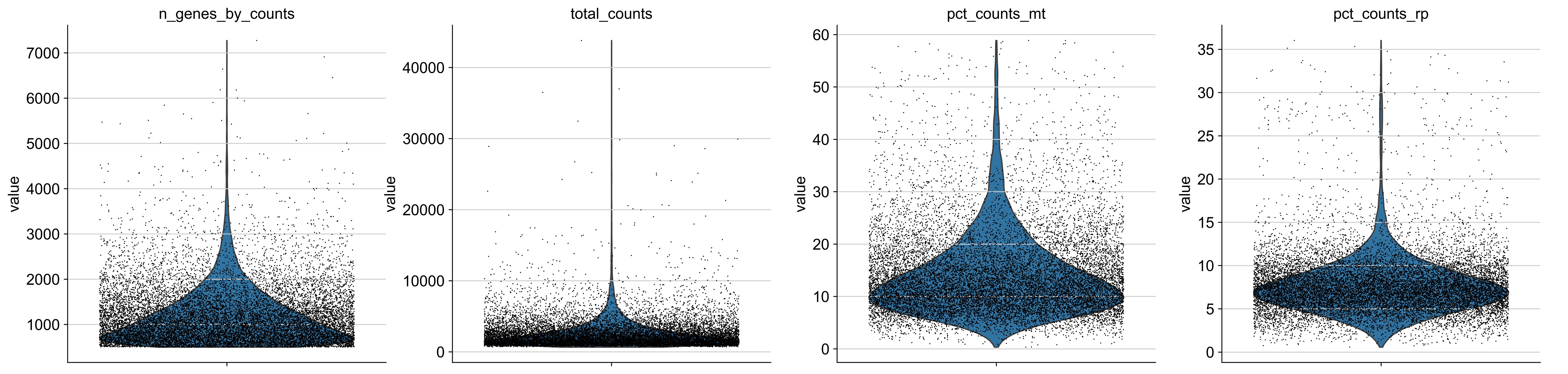
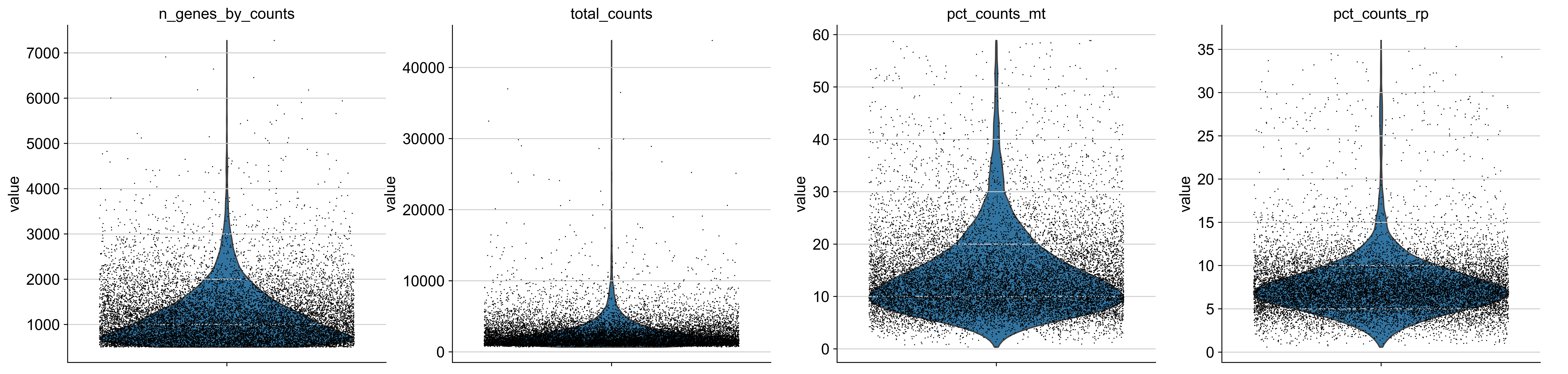
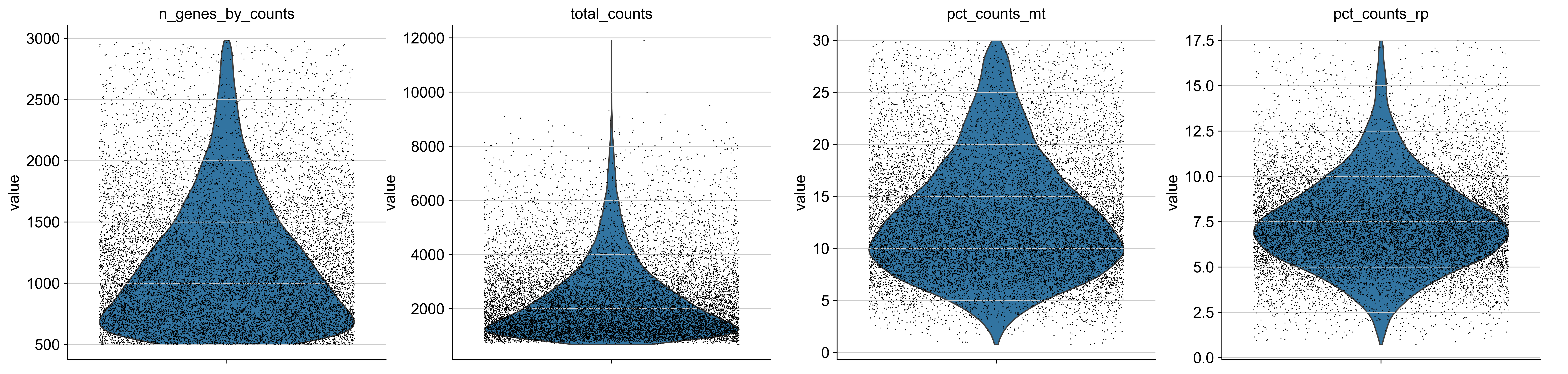
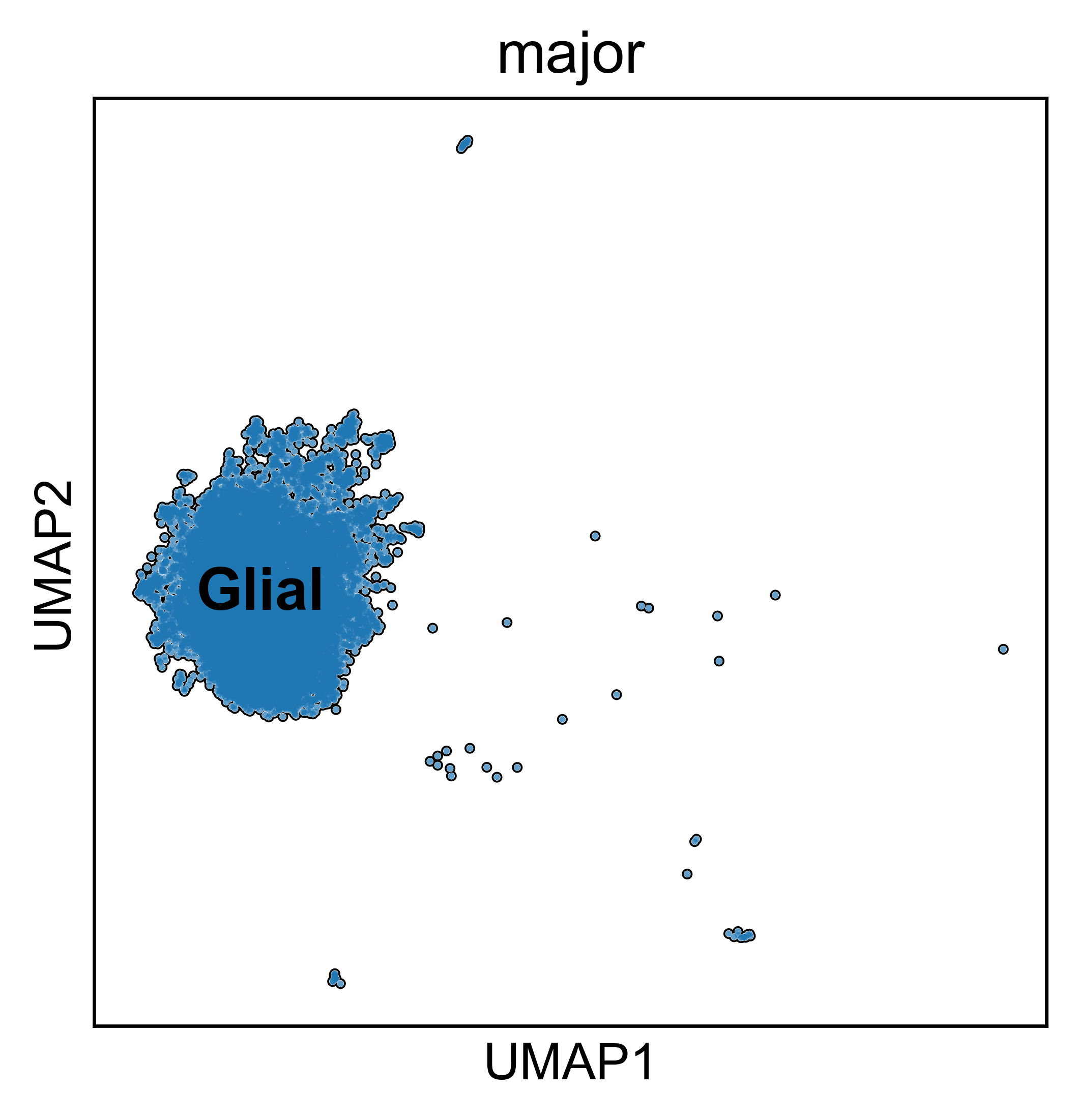
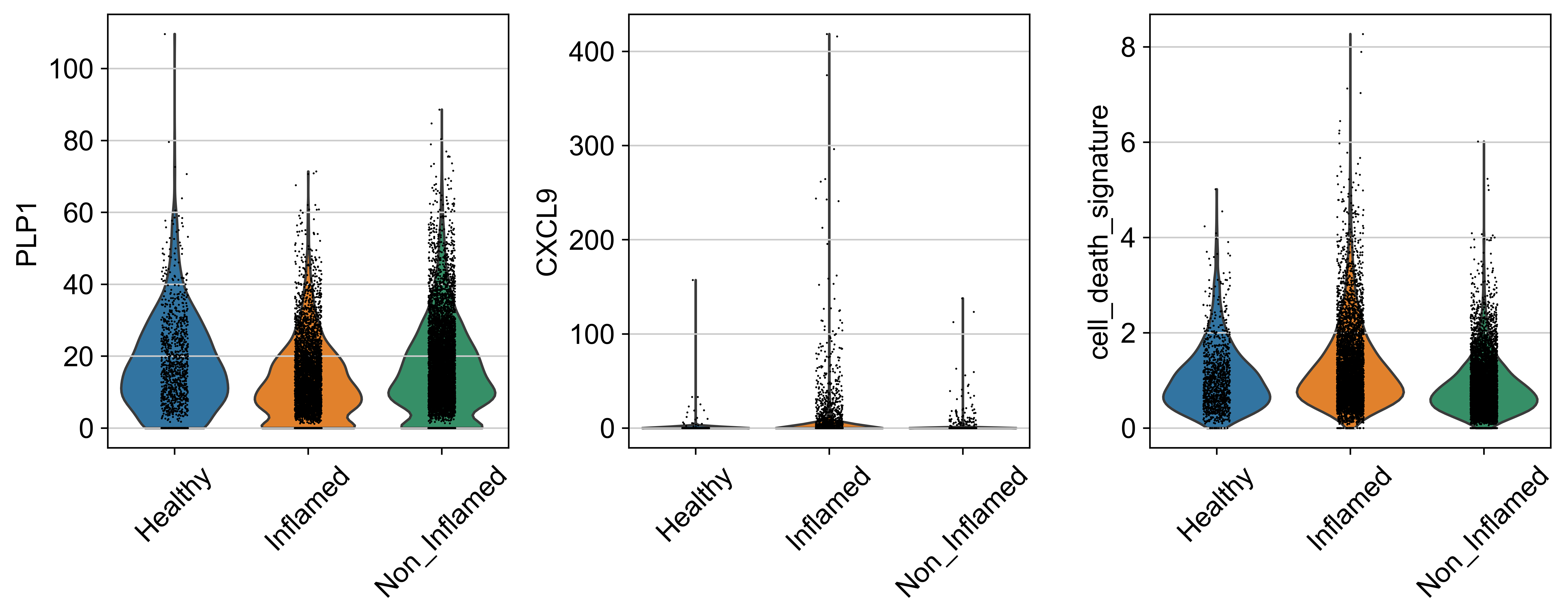
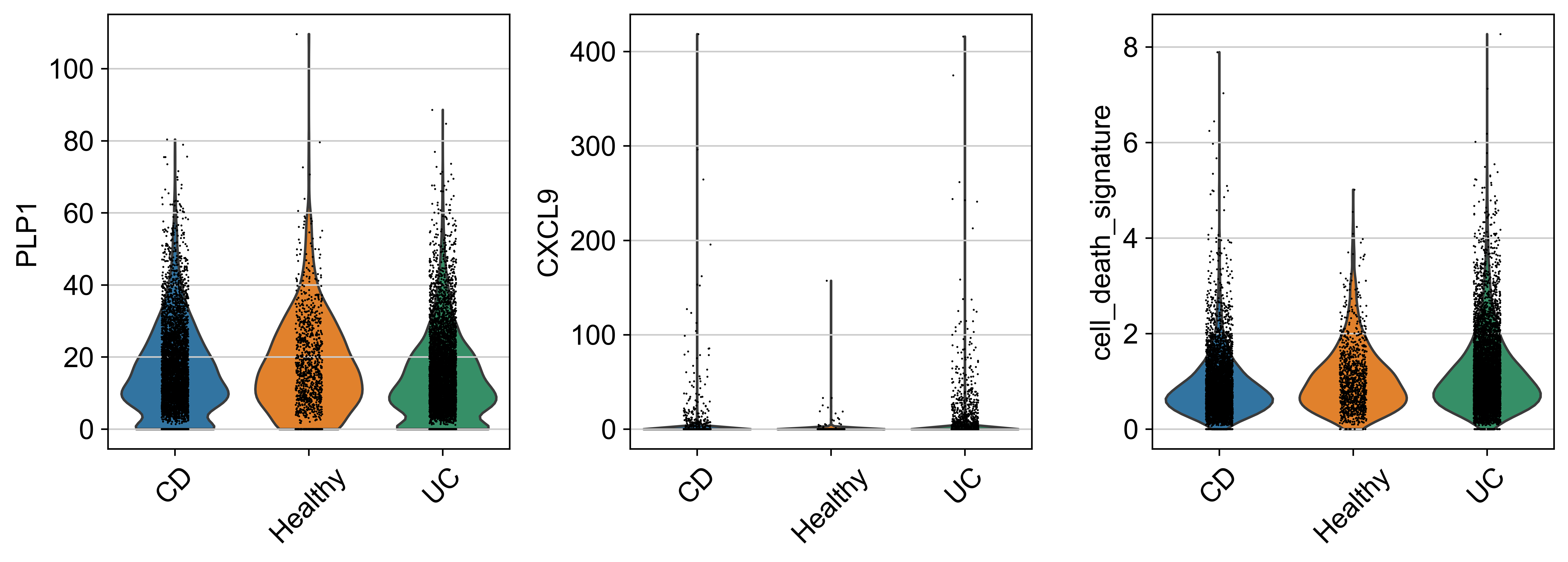
obs: 'sample_id', 'Patient', 'Disease', 'Site', 'Treatment', 'Disease_duration', 'Inflammation', 'Age', 'Gender', 'Ethnicity', 'Inflammation_score', 'Ileum_vs_Colon', 'LibraryType', 'CellsLoaded', 'Match', 'Batch', 'doublet_scores', 'predicted_doublets', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'total_counts_rp', 'pct_counts_rp', 'total_counts_hb', 'pct_counts_hb', 'total_counts_ig', 'pct_counts_ig', 'S_score', 'G2M_score', 'phase', 'cellbarcode', 'diff', 'final_analysis', 'minor', 'major', 'sub_bucket', 'bucket', 'Remission_status'
var: 'feature_types', 'gene_id', 'hb', 'ig', 'rp', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'gene_symbol'
uns: 'final_analysis_colors', 'log1p', 'sample_id_colors'
obsm: 'X_harmony', 'X_pca', 'X_umap'